
Conducting a Human Health Risk Assessment
Planning and Scoping
Even a human health risk assessment starts with a good plan. Before anything though there is a need to make judgments early when planning major risk assessments regarding the purpose, scope, and technical approaches that will be used. To start, risk assessors will typically ask the following questions:
- Who/What/Where is at risk?
- Individual
- General population
- Lifestages such as children, teenagers, pregnant/nursing women
- Population subgroups - highly susceptible (for example, due to asthma, genetics, etc.) and/or highly exposed (for example, based on geographic area, sex, racial or ethnic group, or economic status)
- What is the environmental hazard of concern?
- Chemicals (single or multiple/cumulative risk)
- Radiation
- Physical (dust, heat)
- Microbiological or biological
- Nutritional (for example, diet, fitness, or metabolic state)
- Socio-Economic ( for example, access to health care)
- Where do these environmental hazards come from?
- Point sources (for example, smoke or water discharge from a factory; contamination from a Superfund site)
- Non-point sources (for example, automobile exhaust; agricultural runoff)
- Natural sources
- How does exposure occur?
- Pathways (recognizing that one or more may be involved)
- Air
- Surface Water
- Groundwater
- Soil
- Solid Waste
- Food
- Non-food consumer products, pharmaceuticals
- Routes (and related human activities that lead to exposure)
- Ingestion (both food and water)
- Contact with skin
- Inhalation
- Non-dietary ingestion (for example, "hand-to-mouth" behavior)
- Pathways (recognizing that one or more may be involved)
- What does the body do with the environmental hazard and how is this impacted by factors such as age, race, sex, genetics, etc.?)
- Absorption - does the body take up the environmental hazard
- Distribution - does the environmental hazard travel throughout the body or does it stay in one place?
- Metabolism - does the body break down the environmental hazard?
- Excretion - how does the body get rid of it?
- What are the health effects?
- Example of some health effects include cancer, heart disease, liver disease and nerve disease.
- Example of some health effects include cancer, heart disease, liver disease and nerve disease.
- How long does it take for an environmental hazard to cause a toxic effect? Does it matter when in a lifetime exposure occurs?
- How long?
- Acute - right away or within a few hours to a day
- Subchronic - weeks or months (for humans generally less than 10% of their lifespan)
- Chronic - a significant part of a lifetime or a lifetime (for humans at least seven years)
- Intermittent
- Timing
Is there a critical time during a lifetime when a chemical is most toxic (e.g., fetal development, childhood, during aging)?
- How long?
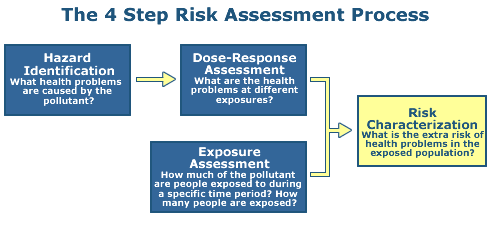
Step 1: Hazard Identification
The objective of Step 1 is to identify the types of adverse health effects that can be caused by exposure to some agent in question, and to characterize the quality and weight of evidence supporting this identification.
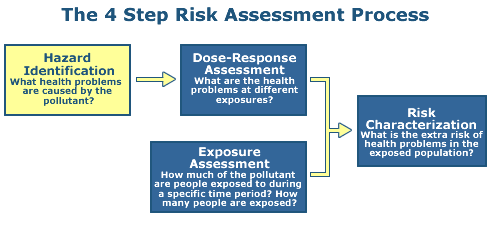
In the case of chemical stressors, the process examines the available scientific data for a given chemical (or group of chemicals) and develops a weight of evidence to characterize the link between the negative effects and the chemical agent.
Exposure to a stressor may generate many different adverse effects in a human: diseases, formation of tumors, reproductive defects, death, or other effects.
Sources of Data
Statistically controlled clinical studies on humans provide the best evidence linking a stressor, often a chemical, to a resulting effect. However, such studies are frequently not available since there are significant ethical concerns associated with human testing of environmental hazards.
Epidemiological studies involve a statistical evaluation of human populations to examine whether there is an association between exposure to a stressor and a human health effect. The advantage of these studies is that they involve humans while their weakness results from generally not having accurate exposure information and the difficulty of teasing out the effects of multiple stressors.
When data from human studies are unavailable, data from animal studies (rats, mice, rabbits, monkeys, dogs, etc) are relied on to draw inference about the potential hazard to humans. Animal studies can be designed, controlled, and conducted to address specific gaps in knowledge, but there are uncertainties associated with extrapolating results from animal subjects to humans.
Key Components of Hazard Identification
A wide variety of studies and analysis are used to support a hazard identification analysis.
- ToxicokineticsThe passage through the body of a toxic agent or its metabolites, usually in an action similar to that of pharmacokinetics. considers how the body absorbs, distributes, metabolizes, and eliminates specific chemicals.
- ToxicodynamicsThe physiological mechanisms by which toxins are absorbed, distributed, metabolized and excreted. focus on the effects that chemicals have on the human body. Models based on these studies can describe mechanisms by which a chemical may impact human health, thus providing insights into the possible effects of a chemical.
When assessing a chemical for potential carcinogenic behavior, current EPA practice is to focus on analysis of a mode of actionDefined as a sequence of key events and processes, starting with interaction of an agent with a cell, proceeding through operational and anatomical changes, and resulting in cancer formation.. Mode of action is a sequence of key events and processes, starting with interaction of an agent and a cell, proceeding through operational and anatomical changes, and resulting in cancer formation. A given agent may work by more than one mode of action, both at different tumor sites as well as at the same site.
Analysis of mode of action is based on physical, chemical, and biological information that helps to explain key events in an agent's influence on tumor development.
A key component of hazard characterization involves evaluating the weight of evidenceWeight of evidence (WOE) category. Based on the quality and adequacy of data on carcinogenicity, EPA places a chemical in one of the following five weight of evidence categories, as specified in 51 FR 33996: A. Carcinogenic to humans; B. Probable carcinogen; B1. Indicates limited human evidence; B2. Indicates sufficient evidence in animals and inadequate or no evidence in humans; C. Possible carcinogen; D. Not classifiable; E. Evidence of non-carcinogenicity regarding a chemical’s potential to cause adverse human health effects. The weight of evidence narrative may include some standard 'descriptors' that signify certain qualitative threshold levels of evidence or confidence have been met, such as 'Carcinogenic to humans' or 'Suggestive evidence of carcinogenic potential'.
Note: More specific information on hazard assessment approaches can be found in the specific risk assessment guidelines relevant for the kind of effect being considered.
Step 2: Dose Response
The objective of Step 2 is to document the relationship between dose and toxic effect.
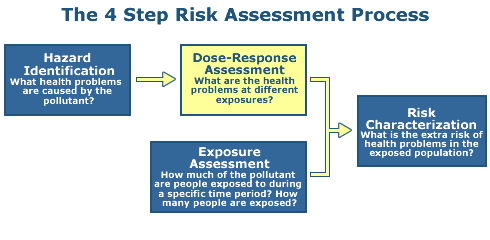
The term "exposure-responseThe relationship between the intensity, frequency, or duration of exposure to a stressor and the intensity, frequency, or duration of the biological response." relationship may be used to describe either a dose-response or a concentration-response, or other specific exposure conditions.
Typically, as the dose increases, the measured response also increases. At low doses there may be no response. At some level of dose the responses begin to occur in a small fraction of the study population or at a low probability rate. Both the dose at which response begin to appear and the rate at which it increases given increasing dose can be variable between different pollutants, individuals, exposure routes, etc.
The shape of the dose-response relationship depends on the agent, the kind of response (tumor, incidence of disease, death, etc), and the experimental subject (human, animal) in question. For example, there may be one relationship for a response such as 'weight loss' and a different relationship for another response such as 'death'. Since it is impractical to study all possible relationships for all possible responses, toxicity research typically focuses on testing for a limited number of adverse effects.
Typically, as the dose increases, the measured response also increases.
Upon considering all available studies, the response (adverse effect), or a measure of response that leads to an adverse effect (known as a ‘precursor’ to the effect), that occurs at the lowest dose is selected as the critical effect for risk assessment. The underlying assumption is that if the critical effect is prevented from occurring, then no other effects of concern will occur.
As with hazard identification, there is frequently a lack of dose-response data available for human subjects. When data are available, they often cover only a portion of the possible range of the dose-response relationship, in which case some extrapolation must be done in order to extrapolate to dose levels that are lower than the range of data obtained from scientific studies. Also, as with hazard identification, animal studies are frequently done to augment the available data.
Studies using animal subjects permit the use of study design to control the number and composition (age, sex, species) of test subjects, the levels of dose tested, and the measurement of specific responses. Use of a designed study typically leads to more meaningful statistical conclusions than does an uncontrolled observational study were additional confounding factors must also be considered for their impact on the conclusions.
However, dose-response relationships observed from animal studies are often at much higher doses that would be anticipated for humans, so must be extrapolated to lower doses, and animal studies must also be extrapolated from that animal species to humans in order to predict the relationship for humans. These extrapolations, among others, introduce uncertainty into the dose-response analysis.
Basic Dose-Response Calculations & Concepts
As a component of the first step of the process discussed below, the scientific information is evaluated for a better biological understanding of how each type of toxicity or response (adverse effect) occurs; the understanding of how the toxicity is caused is called the "mode of action" (which is defined as a sequence of key events and processes, starting with interaction of an agent with a cell, proceeding through operational and anatomical changes, and resulting in the effect, for example, cancer formation).
Based on this mode of action, the Agency determines the nature of the extrapolation used in the second step of the process discussed above, either through non-linear or linear dose-response assessment.
Step 1: Take an assessment of all data that are available or can be gathered through experiments. This is in order to document the dose-response relationship(s) over the range of observed doses (i.e, the doses that are reported in the data collected).
However, frequently this range of observation may not include sufficient data to identify a dose where the adverse effect is not observed (i.e., the dose that is low enough to prevent the effect) in the human population.
Step 2: This consists of extrapolation to estimate the risk (probably of adverse effect) beyond the lower range of available observed data. This is in order to make inferences about the critical region where the dose level begins to cause the adverse effect in the human population.
Non-linear dose-response assessment
Non-linear dose responseA pattern of frequency or severity of biological response that does not vary directly with the amount of dose of an agent. assessment has its origins in the threshold hypothesis, which holds that a range of exposures from zero to some finite value can be tolerated by the organism with essentially no chance of expression of the toxic effect, and the threshold of toxicity is where the effects (or their precursors) begin to occur. It is often prudent to focus on the most sensitive members of the population; therefore, regulatory efforts are generally made to keep exposures below the population threshold, which is defined as the lowest of the thresholds of the individuals within a population.
If the "mode of action" information (discussed above) suggests that the toxicity has a threshold, which is defined as the dose below which no deleterious effect is expected to occur, then type of assessment is referred to by the Agency as a "non-linear" dose-response assessment. The term "nonlinear" is used here in a narrower sense than its usual meaning in the field of mathematics; a nonlinear assessment uses a dose-response relationship whose slope is zero (i.e., no response) at (and perhaps above) a dose of zero.
A No-Observed-Adverse-Effect Level (NOAELThe highest exposure level at which there are no biologically significant increases in the frequency or severity of adverse effect between the exposed population and its appropriate control; some effects may be produced at this level, but they are not considered adverse or precursors of adverse effects.) is the highest exposure level at which no statistically or biologically significant increases are seen in the frequency or severity of adverse effect between the exposed population and its appropriate control population. In an experiment with several NOAELs, the regulatory focus is normally on the highest one, leading to the common usage of the term NOAEL as the highest experimentally determined dose without a statistically or biologically significant adverse effect. In cases in which a NOAEL has not been demonstrated experimentally, the term "lowest-observed-adverse-effect level (LOAEL)" is used, which this is the lowest dose tested.
Mathematical modeling, which can incorporate more than one effect level (i.e., evaluates more data than a single NOAEL or LOAEL), is sometimes used to develop an alternative to a NOAEL known as a Benchmark Dose (BMD) or Benchmark Dose Lower-confidence Limit (BMDL). In developing the BMDL, a predetermined change in the response rate of an adverse effect (called the benchmark response or BMR; generally in the range of 1 to 10% depending on the power of a toxicity study) is selected, and the BMDL is a statistical lower confidence limit on the dose that produces the selected response. When the non-linear approach is applied, the LOAEL, NOAEL, or BMDL is used as the point of departure for extrapolation to lower doses.
The reference dose (RfDAn estimate (with uncertainty spanning perhaps an order of magnitude) of a daily oral exposure to the human population (including sensitive subgroups) that is likely to be without an appreciable risk of deleterious effects during a lifetime. It can be derived from a NOAEL, LOAEL, or benchmark dose, with uncertainty factors generally applied to reflect limitations of the data used. Generally used in EPA's non cancer health assessments. [Durations include acute, short-term, sub chronic, and chronic and are defined individually in this glossary].) is an oral or dermal dose derived from the NOAEL, LOAEL or BMDL by application of generally order-of-magnitude uncertainty factors (UFs). These uncertainty factors take into account the variability and uncertainty that are reflected in possible differences between test animals and humans (generally 10-fold or 10x) and variability within the human population (generally another 10x); the UFs are multiplied together: 10 x 10 = 100x.
If a LOAEL is used, another uncertainty factor, generally 10x, is also used. In the absence of key toxicity data (duration or key effects), an extra uncertainty factor(s) may also be employed. Sometimes a partial UF is applied instead of the default value of 10x, and this value can be less than or greater than the default. Often the partial value is ½ log unit (the square root of 10) or 3.16 (rounded to 3-fold in risk assessment). Note, that when two UFs derived from ½ log units are multiplied together (3 x 3) the result is a 10 (equal to the full UF from which the two partial factors were derived).
Thus, the RfD is determined by use of the following equation: RfD = NOAEL (or LOAEL or BMDL) / UFs
In general, the RfD is defined as an estimate (with uncertainty spanning perhaps an order of magnitude) of a daily oral exposure to the human population (including sensitive groups, such as asthmatics, or life stages, such as children or the elderly) that is likely to be without an appreciable risk of deleterious effects during a lifetime.
The RfD is generally expressed in units of milligrams per kilogram of bodyweight per day: mg/kg/day.
A similar term, know as reference concentration (RfCAn estimate (with uncertainty spanning perhaps an order of magnitude) of a continuous inhalation exposure to the human population (including sensitive subgroups) that is likely to be without an appreciable risk of deleterious effects during a lifetime. It can be derived from a NOAEL, LOAEL, or benchmark concentration, with uncertainty factors generally applied to reflect limitations of the data used. Generally used in EPA's non cancer health assessments. [Durations include acute, short-term, sub chronic, and chronic and are defined individually in this glossary].), is used to assess inhalation risks, where concentration refers to levels in the air (generally expressed in the units of milligrams agent per cubic meter of air: mg/m3).
- For more information, please see A Review of the Reference Dose and Reference Concentration Processes.
Linear dose-response assessment
If the "mode of action" information (discussed above) suggests that the toxicity does not have a threshold, then this type of assessment is referred to by the Agency as a "linear" dose-response assessment. In the case of carcinogens, if "mode of action" information is insufficient, then linear extrapolation is typically used as the default approach for dose-response assessment.
- For more detailed information, please see EPA’s Guidelines for Carcinogen Risk Assessment.
In this type of assessment, there is theoretically no level of exposure for such a chemical that does not pose a small, but finite, probability of generating a carcinogenic response. The extrapolation phase of this type of assessment does not use UFs; rather, a straight line is drawn from the point of departure for the observed data (typically the BMDL) to the origin (where there is zero dose and zero response).
The slope of this straight line, called the slope factor or cancer slope factor, is use to estimate risk at exposure levels that fall along the line. When linear dose-response is used to assess cancer risk, EPA calculates excess lifetime cancer risk (i.e., probability that an individual will contract cancer over a lifetime) resulting from exposure to a contaminant by considering the degree to which individuals were exposed, as compared to the slope factor.
Thus, the cancer risk is determined by use of the following equation: Cancer Risk = Exposure x Slope Factor
Total cancer risk is calculated by adding the individual cancer risks for each pollutant in each pathway of concern (i.e., inhalation, ingestion, and dermal absorption), then summing the risk for all pathways.
A similar term, know as inhalation unit riskThe upper-bound excess lifetime cancer risk estimated to result from continuous exposure to an agent at a concentration of 1 μg/m3 in air for a lifetime. (IUR), is used to assess inhalation risks, where the exposure-response relationship refers to concentrations in the air.
Note: When there are alternative procedures having significant biological support, the Agency encourages assessments to be performed using these alternative procedures, if feasible, in order to shed light on the uncertainties in the assessment, recognizing that the Agency may decide to give greater weight to one set of procedures than another in a specific assessment or management decision.
Step 3: Exposure Assessment
The objective of Step 3 is to calculate a numerical estimate of exposure or dose.
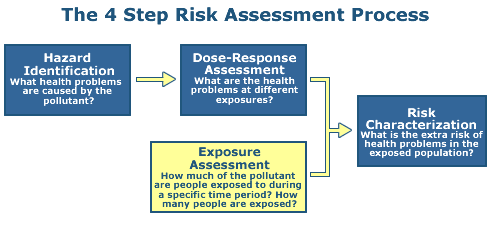
Exposure can be measured directly, but more commonly is estimated indirectly through consideration of measured concentrations in the environment, consideration of models of chemical transport and fate in the environment, and estimates of human intake over time.
Different Kinds of Doses. Exposure assessment considers both the exposure pathway (the course an agent takes from its source to the person(s) being contacted) as well as the exposure route (means of entry of the agent into the body). The exposure route is generally further described as intake (taken in through a body opening, e.g. as eating, drinking, or inhaling) or uptake (absorption through tissues, e.g. through the skin or eye).
EPA defines exposure as 'contact between an agent and the visible exterior of a person (e.g. skin and openings into the body)'.
The applied dose is the amount of agent at the absorption barrier that is available for absorption. The potential dose is the amount of agent that is ingested, inhaled, or applied to the skin. The applied dose may be less than the potential dose if the agent is only partly bioavailable.
The internal dose or absorbed dose is the amount of an agent that has been absorbed and is available for interaction with biologically significant receptors within the human body. Finally, the delivered dose is the amount of agent available for interaction with any specific organ or cell.
Range of Exposure. For any specific agent or site, there is a range of exposures actually experienced by individuals. Some individuals may have a high degree of contact for an extended period (e.g. factory workers exposed to an agent on the job). Other individuals may have a lower degree of contact for a shorter period (e.g. individuals using a recreational site downwind of the factory). EPA policy for exposure assessment requires consideration of a range of possible exposure levels.
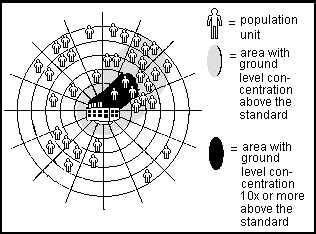
Two common scenarios for possible exposure are "Central Tendency" and "High End". "Central Tendency" exposure is an estimate of the average experienced by the affected population, based on the amount of agent present in the environment and the frequency and duration of exposure.
"High End" exposure is the highest dose estimated to be experienced by some individuals, commonly stated as approximately equal to the 90th percentile exposure category for individuals.
Quantifying Exposure. There are three basic approaches for quantifying exposure. Each approach is based on different data, and has different strengths and weaknesses; using the approaches in combination can greatly strengthen the credibility of an exposure risk assessment.
- Point of Contact Measurement - The exposure can be measured at the point of contact (the outer boundary of the body) while it is taking place, measuring both exposure concentration and time of contact, then integrating them;
- Scenario Evaluation - The exposure can be estimated by separately evaluating the exposure concentration and the time of contact, then combining this information;
- Reconstruction - the exposure can be estimated from dose, which in turn can be reconstructed through internal indicators (biomarkers, body burden, excretion levels, etc) after the exposure has taken place (reconstruction).
- For more information on exposure assessment methods, see the Guidelines for Exposure Assessment.
Step 4: Risk Characterization
The objective of Step 4 is to summarize and integrate information from the proceeding steps of the risk assessment to synthesize an overall conclusion about risk.
In practice, each component of the risk assessment (e.g. hazard assessment, dose-response assessment, exposure assessment) has an individual risk characterization written to carry forward the key findings, assumptions, limitations, and uncertainties. The set of these individual risk characterizations provide the information basis to write an integrative risk characterization analysis.
The final, overall risk characterization thus consists of the individual risk characterizations plus an integrative analysis.
The overall risk characterization informs the risk manager and others about the rationale behind EPA's approach to conducting the risk assessment.
Such as: Why EPA did what it did to assess the risk?
Principles of Conducting Risk Characterizations
A good risk characterization will restate the scope of the assessment, express results clearly, articulate major assumptions and uncertainties, identify reasonable alternative interpretations, and separate scientific conclusions from policy judgments.
EPA's Risk Characterization Policy calls for conducting risk characterizations in a manner that is consistent with the following principles:
- Transparency - The characterization should fully and explicitly disclose the risk assessment methods, default assumptions, logic, rationale, extrapolations, uncertainties, and overall strength of each step in the assessment.
- Clarity - The products from the risk assessment should be readily understood by readers inside and outside of the risk assessment process. Documents should be concise, free of jargon, and should use understandable tables, graphs, and equations as needed.
- Consistency - The risk assessment should be conducted and presented in a manner which is consistent with EPA policy, and consistent with other risk characterizations of similar scope prepared across programs within the EPA.
- Reasonableness - The risk assessment should be based on sound judgment, with methods and assumptions consistent with the current state-of-the-science and conveyed in a manner that is complete and balanced, informative.
These four principles are referred to collectively as TCCR. In order to achieve TCCR in a risk characterization, the same principles need to have been applied in all of the prior steps in the risk assessment which lead up to the risk characterization.
- For more information read the Risk Characterization Handbook.